The Hamartomatous (Mesenchymal) Syndromes
HEREDITARY
5/27/20248 min read
The Syndromes we have already considered, FAP, MAP, PPAP, and SPS, are epithelial syndromes that affect cells lining the colon. They produce adenomas and adenocarcinomas (and serrated polyps). Hamartomas are different. They are benign overgrowths of normal tissue located under the lining of the colon that happen due to mutations of genes in different pathways to those causing the epithelial cancers. There is still loss of growth control of cells in the wall of the colon but the cells overgrow in a non-threatening way. Sometimes an epithelial cancer develops, due to the effect that the genetic mutations can have on epithelial (lining) cells. Although this is not so common, the cancer risk is still higher than average. Therefore we need to keep checking the organs at risk. There are three main syndromes:
Peutz-Jegher’s Polyposis (PJP)
Juvenile Polyposis Syndrome (JPS)
PTEN Hamartoma Tumor Syndrome (PHTS)
Peutz-Jeghers Syndrome (PJS)
Peutz-Jeghers is a dominantly inherited syndrome of abnormal cell growth due to a germline mutation in the tumor suppressor gene STK11(or LKB1). The protein coded for by the gene is involved in cell growth control through its interactions with p53 and PTEN, and also has a role in repair of DNA damage due to ultraviolet radiation. The effect of STK11 mutation on the functions of p53 and PTEN is reflected in the wide range of tumors seen in the syndrome.
Diagnosis
PJS is rare, approximately 1/10 the frequency of FAP. Mutations in STK11 are found in about 69% of people with clinical PJS. Diagnostic criteria are: 1. ≥2 Peutz-Jeghers polyps 2. Any Peutz-Jeghers polyps and a family history of PJS 3. Freckles on the lips and in the cheek mucosa plus a family history of PJS 4. Freckles on the lips and in the cheek mucosa plus any Peutz-Jeghers polyps
Presentation
The primary expression of the mutation is in the GI tract with hamartomatous polyps in the small intestine, stomach and colon. 50% of affected patients present with small bowel obstructive symptoms in the first 10 years of life. Other organs are affected with a range of tumors as shown.
Pathology
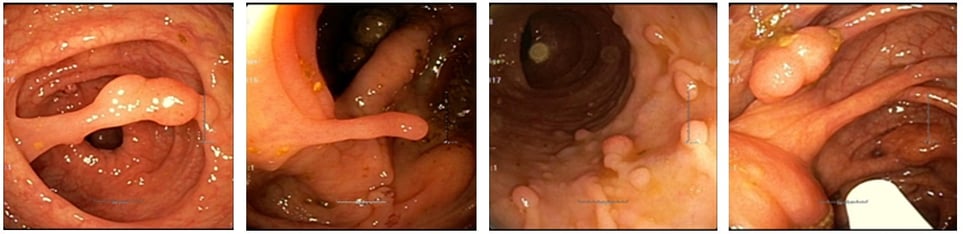
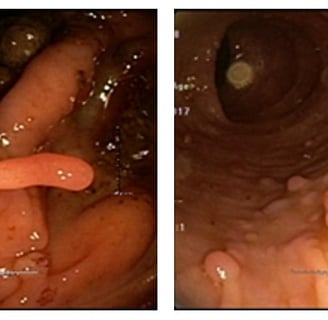
Peutz-Jeghers polyps are hamartomas of the muscularis mucosa. This causes branching muscle fibers in the polyps. The epithelium of the polyps is normal but is prone to later develop cancer. Endoscopically the polyps have long stalks.
Peutz-Jeghers Syndrome Surveillance
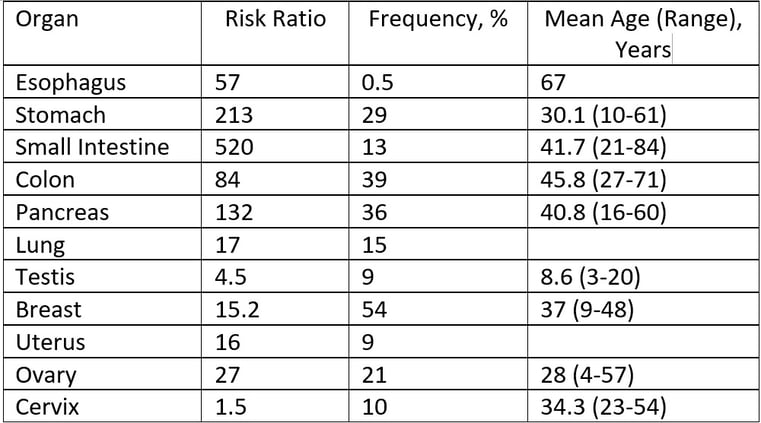
Start 5-10 years before the youngest age of diagnosis of a cancer in the family if this is younger
*Jelsig AM, Qvist N, Brusgaard K, Nielsen CB, Hansen TP, Ousager LB (July 2014). "Hamartomatous polyposis syndromes: a review". Orphanet Journal of Rare Diseases. 9: 101.
Bannayan-Riley-Ruvalcaba Syndrome (BRRS)
Bannayan-Riley-Ruvalcaba Syndrome is a rare hereditary growth disorder characterized by lipomatosis, macrocephaly, a speckled penis, intestinal hamartomas skeletal abnormalities and developmental delay/autism spectrum disorder. It is dominantly inherited and 60% of cases carry a germline pathogenic variant in PTEN. There is overlap between the clinical features of BRRS and Cowden Syndrome, although BRRS may present in childhood.
Gastrointestinal hamartomatous polyposis may present earlier than with Cowden Syndrome. Diarrhea and rectal bleeding are indications for colonoscopy.
Thyroid ultrasound should begin at age 7 and evaluation for autism spectrum disorder should be considered.
In older cases, yearly skin check should start at 18 years of age, colonoscopy and renal ultrasound at 35 to 40, with yearly breast screening, and transvaginal ultrasound from 35.
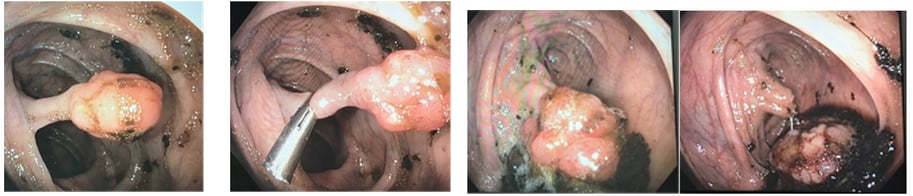
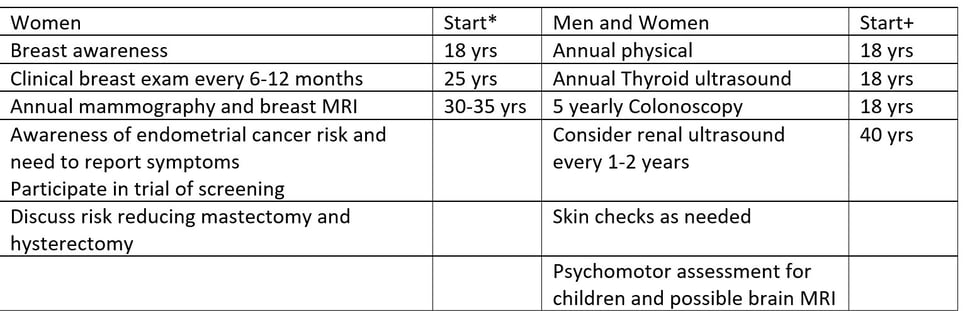
Colectomy for Cowden Syndrome. Note the hypertrophic mesentery and epiploicae
Resect the colon if colonoscopy cannot control the dangerous polyps.
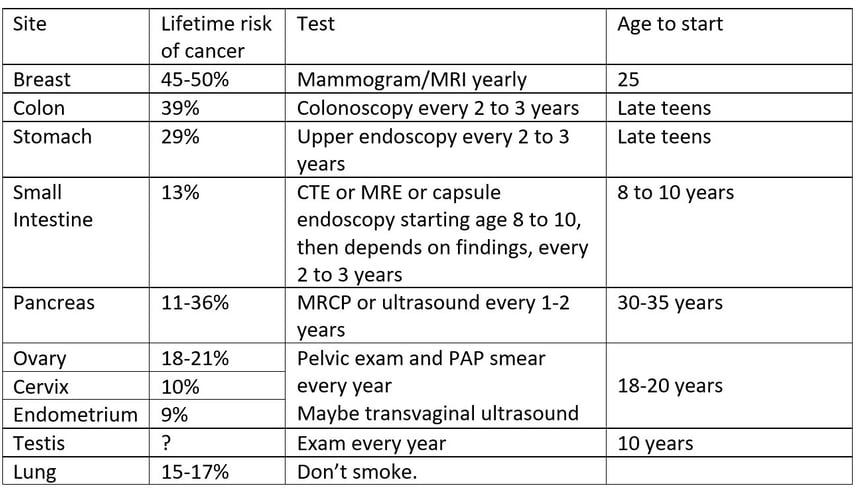
Surveillance for Cowden Patients (adapted from NCCN*)
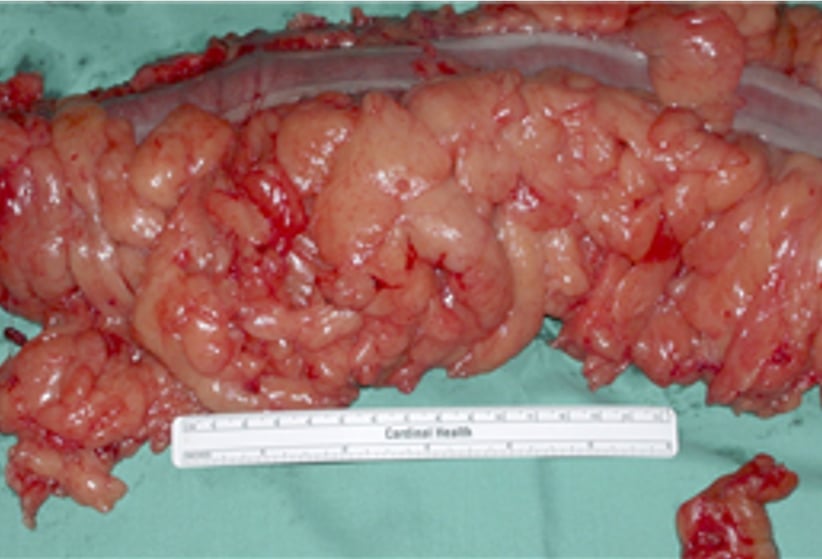
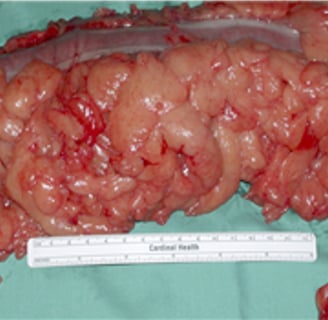
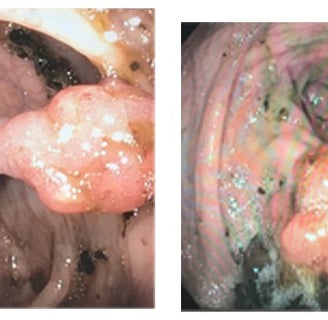
Multiple polyps of various histology in Cowden Syndrome
Investigation and Therapy
Investigation of the gastrointestinal tract uses endoscopy and radiology. An EGD and a push or balloon enteroscopy can evaluate the stomach and small intestine and remove some polyps. A colonoscopy can evaluate and treat. A video capsule enteroscopy, or a CT or MR enterography can also evaluate but not treat.
Treatment of small bowel obstruction by polyps or an intussuscepting polyp may need surgery. Just resecting the symptomatic polyps places the patient at risk of subsequent surgeries for recurrent obstruction. The “Clean Sweep” surgery uses combined surgery and enteroscopy to remove all polyps regardless of size, minimizing the chances of another operation.
Removing colonic Peutz-Jeghers Polyps. They tend to bleed so clip the stalk.
Juvenile Polyposis Syndrome (JPS)
Juvenile Polyposis Syndrome (JPS) is a dominantly inherited syndrome of abnormal cell growth due to a germline mutation in the tumor suppressor genes SMAD4 and BMPR1A. The proteins coded for by the gene are involved in the key growth controlling TGF signal transduction pathway. SMAD4 is the central mediator for the TGF pathway and promotes tumors initiated by other gene mutations. It also coordinates TGF pathway with other key pathways including wnt/β catenin and MAPK/ERK. BMPR1A suppresses wnt signaling and has a role in reducing angiogenesis.
Diagnosis
JPS is rare, approximately 1/100,000 people. About 55% of affected patients will have a mutation (28% BMPR1A, 27% SMAD4)
Diagnostic criteria are: 1. more than five juvenile polyps of the colon or rectum; 2. juvenile polyps in other parts of the gastrointestinal tract; or 3. any number of juvenile polyps and one or more affected family members.
Presentation
Solitary Juvenile polyps are the commonest polyp in children. However patients fulfilling one of the criteria need genetic testing. The presentation varies in severity with SMAD4 carriers have more severe colonic polyposis, and gastric polyposis. BMPR1A carriers tend not to have gastric involvement and have a mild colorectal phenotype.
Patients with multiple colorectal polyps can present with rectal bleeding and diarrhea. The diagnosis is made on colonoscopy. This leads to genetic testing. If the testing is negative the patient is still treated as if they have JPS. Patients undergo upper endoscopy and are screened for the present of hereditary hemorrhagic telangiectasia.
Hereditary Hemorrhagic Telangiectasia (HHT)
HHT is a dominantly inherited condition where small malformations of blood vessels (telangiectases) form in the skin and the lining of the gastrointestinal tract. They can causes nose bleeds and bleeding from the bowel.
Arteriovenous malformations (AVMs) can be found in larger organs like the lungs, liver and brain. They allow blood to bypass the capillaries in the organs, going straight from artery to vein. Lung AVMs predispose to emboli and strokes. Liver AVMs predispose to heart failure and portal hypertension. About 25 of all cases of HHT are due to mutations in SMAD4. However almost all patients with SMAD4 mutations and JPS will have HHT. Its important to screen for the AVMs as they can cause problems during endoscopy and surgery.
Surveillance
For gene carriers, or asymptomatic relatives of affected patients, colonoscopy and EGD begin in the mid teens. Polyps are removed and the frequency of colonoscopy drops to yearly. If no polyps are found, the interval is 3 years.
Treatment
For patients with mild polyposis, endoscopic control may be possible with yearly examinations. For severe or symptomatic polyposis surgery is considered. This can be total proctocolectomy with ileostomy, ileal pouch- anal anastomosis, or colectomy with ileorectal anastomosis. SMAD4 mutation carriers may have severe enough gastric polyposis to need a gastrectomy.
The polyposis can be very aggressive, affecting the rectum after ileorectal anastomosis, and even the pouch after ileal pouch-anal anastomosis. Juvenile polyps may regress with sulindac.
PTEN Hamartoma Tumor Syndrome (PHTS)
PTEN Hamartoma Tumor Syndrome (PTHS) refers to a small group of dominantly inherited growth disorder syndromes that include Cowden’s Syndrome, Bannayan Ruvlcaba Riley Syndrome, and proteus Syndrome, and are all associated with germline mutations in PTEN.
PTEN is a tumor suppressor gene that is mutated in many human cancers. It is most commonly mutated in prostate cancer. PTEN has an important role in controlling the Akt signaling pathway, which regulates cell growth, cell survival and cell migration. Its protein also interacts with p53, in its involvement in the response to DNA damage. Germline mutations in PTEN cause three overlapping syndromes of growth disorders in multiple tissues. It was originally thought that there was no increased risk for gastrointestinal tumors but this has now changed. Patients with PHTS are at increased risk for colorectal polyps and cancer.
Cowden Syndrome
a rare, autosomal dominant, familial cancer syndrome characterised by hamartomas, acral keratosis, multiple smooth facial papules, and multiple oral papillomas.
Diagnostic Criteria for Cowden Syndrome
A scoring system (which can be found online) that takes into account phenotype and age at diagnosis has been developed. It allows input of clinical information on an individual suspected of having CS/CLS and subsequently generates the prior probability of finding a PTEN pathogenic variant.
In adults, a clinical threshold score of ten or more leads to a recommendation for referral to a genetics professional to consider PHTS.
In children, macrocephaly and one or more of the following leads to the consideration of PHTS:
Autism or developmental delay
Dermatologic features including lipomas, trichilemmomas, oral papillomas, or penile freckling
Vascular features, such as arteriovenous malformations or hemangiomas
Gastrointestinal polyps
Pediatric-onset thyroid cancer or germ cell tumors
Testing for PTEN mutations:
Only 25% of patients meeting the criteria for the diagnosis of Cowden Syndrome will have a pathogenic variant found in the germline, and for Cowden-like patients (criteria minus one) the percent positive is only 5%. This limits the ability of genetic testing to triage the family. have a pathogenic variant found in the germline, and for Cowden-like patients (criteria minus one) the percent positive is only 5%. This limits the ability of genetic testing to triage the family.
Diagnostic Criteria for Cowden Syndrome
(Tan MH, Mester J, Peterson C, Yang Y, Chen JL, Rybicki LA, Milas K, Pederson H, Remzi B, Orloff MS, Eng C. A clinical scoring system for selection of patients for PTEN mutation testing is proposed on the basis of a prospective study of 3042 probands. Am J Hum Genet. 2011 Jan 7;88(1):42-56)
Pathognomonic criteria
Adult Lhermitte-Duclos disease, defined as the presence of a cerebellar dysplastic gangliocytoma [Zhou et al 2003a]
Mucocutaneous lesions:
Trichilemmomas (facial)
Acral keratoses
Papillomatous lesions
Mucosal lesions
Major criteria
Breast cancer
Epithelial thyroid cancer (non-medullary), especially follicular thyroid cancer
Macrocephaly (occipital frontal circumference ≥97th percentile)
Endometrial carcinoma
Minor criteria
Other thyroid lesions (e.g., adenoma, multinodular goiter)
Intellectual disability (IQ ≤75)
Hamartomatous intestinal polyps
Fibrocystic disease of the breast
Lipomas
Fibromas
Genitourinary tumors (especially renal cell carcinoma)
Genitourinary malformation
Uterine fibroids
A clinical diagnosis of CS is established if an individual meets any one of the following criteria:
Pathognomonic mucocutaneous lesions including one of the following:
Six or more facial papules, of which three or more must be trichilemmomas
Cutaneous facial papules and oral mucosal papillomatosis
Oral mucosal papillomatosis and acral keratoses
Six or more palmoplantar keratoses
Two or more major criteria
One major and three or more minor criteria
The Large Bowel in Cowden Syndrome
Most (95%) PTEN carriers have colorectal polyps. 13% develop adenocarcinoma
The following lesions can be found:
Hamartomas
Serrated polyps
Ganglioneuromas
Adenomas
Juvenile (Inflammatory) polyps
Lipomas
Adenocarcinomas
Upper GI Polyps
Hamartomas,
Fundic gland polyps
Ganglioneuromas
Adenomas
Inflammatory polyps