Molecular Genetics of Colorectal Carcinogenesis
HEREDITARY
5/28/202411 min read
Abstract
An understanding of the molecular pathways by which colorectal neoplasia develops is foundational to an understanding of the clinical aspects of the disease. In this chapter the basics of carcinogenesis in the colon are explained. The nature of DNA and the genetic code are revealed and leads on to a discussion of DNA repair and its relevance to cancer. The chapter then focuses on the genes that regulate cell growth, tumor suppressor genes and proto-oncogenes. Pathogenic variants in these gene are directly responsible for the loss of growth regulation that leads to tumors. Bert Vogelstein has been central in delineating the molecular pathways to colorectal cancer. In 1987 he described the adenoma-carcinoma sequence and in 2015 simplified the concept into one where as few as three driver gene variants are necessary for cancer to develop. Since then a second molecular pathway to colorectal cancer has been described, involving BRAF variants and DNA hypermethylation., This is the serrated pathway leading to cancers with the CpG Island Methylator Phenotype (CIMP). Finally the molecular basis of the hereditary syndromes is discussed, separating them into syndromes manifesting epithelial tumors (adenomas, serrated polyps) and those with sub-epithelial based tumors (hamartomas). Adenomatous polyposes and Lynch syndrome feature germline pathogenic variants in gene with important roles in DNA repair or chromosomal separation. Hamartomous polyposes are associated with perturbations of other pathways regulating cells growth that affect components of mesenchymal and endodermal tissues.
Introduction
Colorectal cancer is a genetic disease. It occurs because of failure of the regulatory pathways that control cell growth, division and differentiation. These regulatory pathways are very important in the colon because the rate of stem cell division is high. The epithelium of the large intestine is renewed every 5 to 6 days, as colonocytes produced by stem cells in the base of the crypts move up the crypt wall towards the lumen where they are shed into the stool. The high rate of stem cell division increases the chances of errors in DNA replication and when these errors occur in genes that are part of growth regulation pathways, the affected clone has a growth and survival advantage over neighboring, normally regulated clones. Subsequent cell divisions in the affected clones produce more errors in increasing numbers of genes, worsening the degree of dysplasia in the affected clone until the cells acquire the ability to invade and metastasize. Cells with this ability are malignant. This chapter summarizes current knowledge about the genetic origins of colorectal cancer. We begin with a simplified description of the basics of cancer genetics as it is fundamental to an understanding of these origins.
DNA and the Genetic Code
DNA is an extraordinarily long molecule consisting of a series of nucleotide bases [adenine (A), guanine (G), thymine (T), cytosine(C)] that are arranged in two strands configured as a double helix. The DNA strands are linked by hydrogen bonds between complementary nucleotides: A to T and G to C. The sequence of nucleotides codes for amino acids, the basic units of protein molecules. DNA holds the codes for all proteins, and proteins are the functional units of human
biology. Because cancer is a disease of cell growth, differentiation and death, the genes coding for proteins that regulate these cellular events are particularly relevant to carcinogenesis.
A gene is a segment of DNA that holds the code for a particular protein. There are approximately 20,000 genes in the human genome, making up about 1% of the DNA molecule. A error in the DNA sequence that is uncorrected creates a variant (mutation). An error that causes an abnormally functioning protein that leads to disease is a pathogenic variant. Colorectal cancer develops due to an accumulation of pathogenic variants in genes that control cell growth, differentiation and death.
DNA Repair and Cancer
DNA is constantly being damaged by the metabolic and environmental exposures that are a part of life. These include the oxidative effects of metabolism and the mutagenesis of ultraviolet rays, viruses, chemicals and toxins. DNA damage is recognized by enzymes and repaired as long as the complementary strand is normal. However, if damaged DNA is transmitted through cell division to the next generation of cells it becomes a permanent variant in the base sequence of that cell’s lineage. Some variants are harmless due to redundancy in the genetic code. A normally functioning protein is produced despite their presence. Others are harmful (deleterious) because they alter protein structure and function.
DNA is most vulnerable during cell division, where the strands separate and form the templates for new molecules in daughter cells. The more rapidly the cells divide, the higher the risk for DNA damage. Much of this damage is stochastic (occurring by chance) while some is encouraged by carcinogens. However, the fidelity of DNA replication is closely protected by a number of DNA repair mechanisms. These include mismatch repair, base excision repair, nucleotide excision repair, double strand break repair, and translesion synthesis. DNA repair can fail if the damage is too severe or if the repair mechanisms themselves are faulty. Failure of DNA repair leads to senescence, apoptosis, or creation of variants. Malignancy arises when cells survive with damaged DNA and pursue a Darwinian course, where “survival of the fittest” promotes increasingly dysregulated growth.
Genes and Regulation of Cell Growth
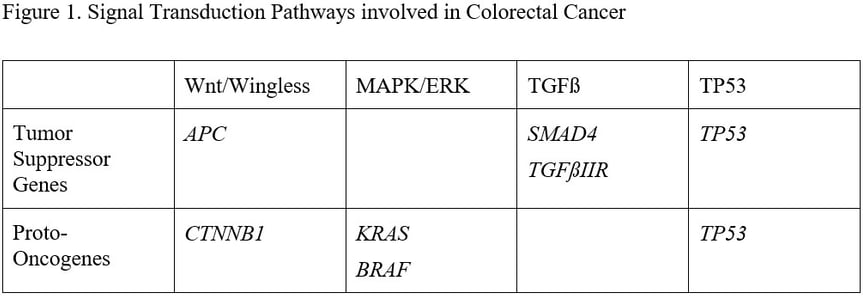
Genes that produce proteins regulating cell growth are tumor suppressor genes (TSG, the protein inhibits cell growth) and proto-oncogenes (the proteins stimulate cell growth). Loss of function of a TSG and gain of function of a proto-oncogene cause abnormally enhanced cell growth. TSG and proto-oncogenes are generally organized into signal transduction pathways, which take a growth-related signal from the cell surface and transmit it through the cytoplasm to the nucleus. DNA is stored in the nucleus and a growth signal causes inactive growth regulating genes to be activated. The pathways involved in colorectal carcinogenesis and the key genes in each pathway are shown in Figure 1.
Genes and Cancer
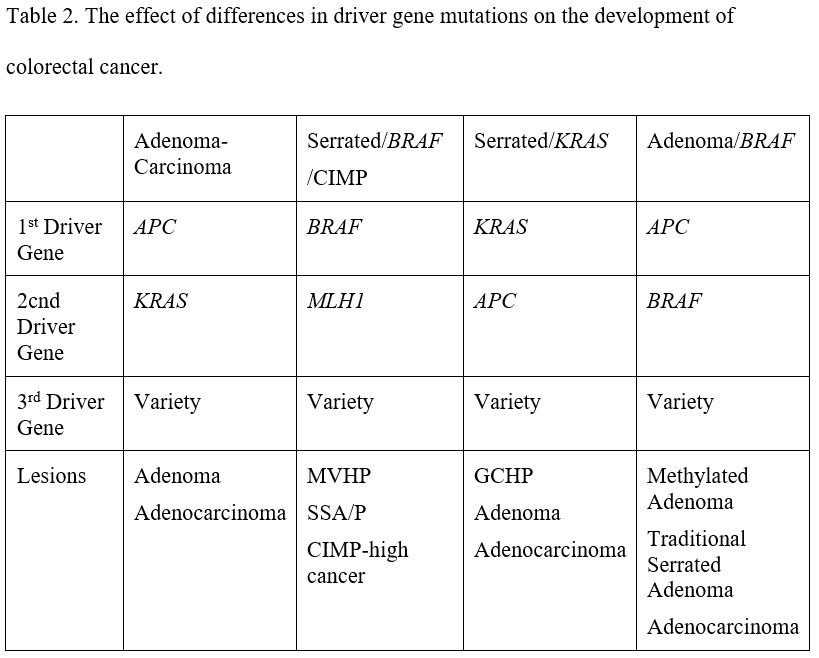
In any cancer there are thousands of genes that contain pathogenic variants. Almost all of these variants are due to generalized cellular instability resulting from the failure of growth control in the cancer clones. These are “passenger” variants. A few genes develop pathogenic variants that drive carcinogenesis. These are the “driver” genes. Vogelstein and Tomasetti suggest that variants in only three driver genes are needed to produce a cancer in a clone of colonocytes. The genes involved and the order in which they are acquired determine the type of neoplasm that develops. There are essentially two main histologic pathways by which sporadic colorectal cancer develops: adenoma to carcinoma, and serrated polyp to carcinoma.
The Adenoma-Carcinoma Sequence
In 1988 Bert Vogelstein described a sequence of genetic changes that correlated with a histologic sequence of neoplasia. The initial genetic event was described as allelic loss in chromosome 5, later shown to be due to loss of APC, a key tumor suppressor gene in the wnt/wingless signal transduction pathway. The next event was an activating variant in KRAS, a protooncogene central to the MAPK/ERK signal transduction pathway. Other allelic loss was also seen in chromosomes 18 (later shown to be due to loss of SMAD4) and 17 (later shown to be P53). SMAD4 is a tumor suppressor gene in the TGF beta signal transduction pathway and P53 is critical to control of cell cycle arrest, DNA damage control and apoptosis. Integration of Vogelstein’s adenoma-carcinoma sequence with his more recent “three strikes and you’re out” concept about colorectal carcinogenesis suggests that if APC is the first driver gene to suffer a pathogenic variant, then carcinogenesis begins with an adenoma. Chromosomal instability is a feature of an adenoma, and is a result of APC inactivation. The multiple chromosomal abnormalities that are features of chromosomal instability lead to loss of heterozygosity of multiple genes, and are detectable as aneuploidy. The adenoma enlarges and becomes increasingly dysplastic with the development of a variant in a second driver gene (KRAS) and develops into a cancer with a third (one of several but including SMAD4 and TP53).
The serrated polyp to adenocarcinoma pathway
About 18% of sporadic colorectal cancers arise through the serrated pathway, which begins with a pathogenic variant in the proto-oncogenes KRAS or BRAF. This activates the MAPK/ERK pathway, causing a failure of apoptosis that produces a buildup of colonocytes within the colonic crypts. This is apparent as the epithelial serrations that characterize serrated polyps. The serrated phenotype varies according to the driver gene, with KRAS variants associated with left sided goblet cell hyperplastic polyps and BRAF variants with right sided microvesicular hyperplastic polyps. In addition, BRAF variants are associated with a carcinogenic pattern of DNA hypermethylation, predominantly on the right side of the colon. One of the genes that can be methylated is MLH1. MLH1 is a DNA mismatch repair gene that, when mutated in the germline, causes Lynch syndrome. However, when it is methylated in a serrated polyp it promotes cytologic dysplasia within the polyp. BRAF-induced DNA hypermethylation and loss of MLH1 expression ultimately lead to colorectal cancers that have a CpG Island Methylator Phenotype (CIMP). Back on the left side of the colon, the KRAS variants that produce goblet cell hyperplastic polyps are associated with a different, less carcinogenic pattern of methylation. There can be overlap between the adenoma-carcinoma pathway and the serrated pathway. When KRAS variants combine in a clone of colonocytes with APC variants, aggressive neoplasms can result through an enhanced adenoma-carcinoma sequence, and the hypermethylation characteristic of CIMP can produce aggression in adenomas. This overlap is shown in Table 2.
Epigenetics: DNA Methylation
Epigenetics refers to the normal mechanisms by which gene expression is regulated without changing the structure of genes. Mechanisms involved in epigenetic modification of gene expression include paramutation, imprinting, histone modification, X chromosome inactivation, and bookmarking. In colorectal carcinogenesis, the relevant epigenetic event is hypermethylation. Here, methyl groups are added or removed from the DNA by DNA methyltransferases. In general, adding methyl groups reduces gene expression while removing methyl groups enhances gene expression. DNA methylation is reversible, and this is a common way that gene expression is regulated in normal tissues. In particular it is methylation of the promoter region of the gene that controls gene expression. Methylation occurs on the cytosine nucleotide base, when it is part of a Cytosine/phosphate/Guanine (CpG) dinucleotide. This dinucleotide is generally under-represented in human DNA, except in the promoter regions of genes. Here there are clusters (islands) of CpG dinucleotides that are analogous to a switch controlling gene expression. However, when the CpG islands of tumor suppressor genes are excessively or inappropriately methylated the effect is to inappropriately reduce or eliminate gene expression. When methylation levels are abnormally low, some genes will be inappropriately expressed. Both hypo and hypermethylation are implicated in carcinogenesis, with global DNA hypomethylation described by Vogelstein in 1983 and hypermethylation in CpG islands by Issa in 1995. Issa coined the term “CpG Island Methylator Phenotype (CIMP)”, associated with a subset of colorectal cancers defined by a phenotype of right sided location and microsatellite instability. He found that DNA methylation in the colonic mucosa increases with age, and is affected by a variety of carcinogens (e.g. smoking). More recently a connection has been established between deleterious variants in BRAF and KRAS and patterns of hypermethylation in the colorectal mucosa. The pattern of hypermethylation associated with BRAF is CIMP and BRAF variants underlie the more aggressive sessile serrated polyps/adenomas that occur on the right side of the colon with increasing frequency as people age. The most severe expression of this genotype is sessile serrated polyposis.
The Genetics of Hereditary Colorectal Cancer
Most colorectal cancers are sporadic, arising by chance from a single colonic crypt where a clone from a single stem cell has been affected by an accumulation of growth deregulating variants. The first driver mutation either arises by chance or though the effect of environmental factors. Hereditary cancers are different. Every cell in an affected patient’s body carries the same critical pathogenic variant that was inherited from their parent’s germline or arose de novo at conception. In theory therefore, every stem cell has the opportunity to generate a clone destined for malignancy, and to generate it early. Inherited cancers therefore tend to be multifocal and to develop in the young.
The histology of the growths that are features of each syndrome are a way of classifying the syndrome, and are also relevant to the underlying carcinogenesis. Hereditary colorectal cancer syndromes can be divided into those producing adenomas and those that produce hamartomas. One of the most important characteristics of adenoma producing syndromes is the way in which the causative germline variants set up a “variant phenotype”, amplifying the variant signal many times over. This is because the causative germline variants involved are almost always important components of DNA repair, so that the consequences of pathogenic variants potentially affect the whole genome. The classic example of this is Lynch syndrome, where a pathogenic variant in a single DNA mismatch repair gene produces variants in thousands of genes via microsatellite instability. MUTYH and NTHL1 associated polyposes also feature variant amplification as defective base excision repair gives rise to C>T transitions (NAP) and G>T transversions (MAP) in multiple genes. Furthermore, pathogenic variants in POLD1 and POLE cause Polymerase proof-reading associated polyposis by allowing errors in DNA replication in multiple genes. A germline variant in APC does not cause variant amplification in the same way as in DNA repair genes, but its effects are amplified by the important role of APC in chromosome segregation. Not only do pathologenic APC variants cause ß catenin-induced activation of growth pathways but they also produce chromosomal instability, with loss of heterozygosity in multiple genes.
The hamartomatous syndromes are different to the neoplastic syndromes because the genetic abnormalities causing them do not directly affect the colorectal epithelium. The growth dysregulation occurs via different pathways that are active in other components of the large bowel, such as the muscularis mucosae in Peutz-Jeghers polyposis and the lamina propria in juvenile polyposis and PTEN-hamartoma tumor syndromes. Because these subepithelial tissues are not in contact with luminal carcinogens there are lower rates of colorectal cancer compared to the other syndromes. However, cancer rates in the colorectum are still increased due to the general instability resulting from the germline variants. Table 3 summarizes the genotypes of hereditary colorectal cancer syndromes.
Table 3. Genotypes of common hereditary colorectal cancer syndromes
Summary
Understanding the origin of colorectal cancer means coming to grips with genetic pathways controlling cell growth and DNA repair, and the ways that disturbances of those pathways influence regulation of colonocyte growth. Hereditary colorectal cancer syndromes arise when a harmful driver gene variant is inherited, creating a variant amplification system that is clinically obvious as multiple tumors arising at an early age. A more detailed description of the genes associated with the syndromes is given in the individual syndrome chapters.
Readings
Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP. Alterations in DNA methylation: a fundamental aspect of neoplasia. Adv Cancer Res. 1998;72:141-96. Review.
Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986 May 15-21;321(6067):209-13.
Feinberg AP, Vogelstein B. Alterations in DNA methylation in human colon neoplasia. Semin Surg Oncol. 1987;3(3):149-51.
Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan RS, Zborowska E, Kinzler KW, Vogelstein B, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science. 1995 Jun 2;268(5215):1336-8.
Ohtani-Fujita, N., Fujita, T., Aoike, A., Osifchin, N. E., Robbins, P. D., and Sakai, T. (1993). CpG methylation inactivates the promoter activity of the human retinoblastoma tumor-suppressor gene. Oncogene 8, 1063-1067.
Tomasetti C, Li L, Vogelstein B. Stem cell divisions, somatic mutations, cancer etiology, and cancer prevention. Science. 2017 Mar 24;355(6331):1330-1334.
Tomasetti C, Durrett R, Kimmel M, Lambert A, Parmigiani G, Zauber A, Vogelstein B. Role of stem-cell divisions in cancer risk. Nature. 2017 Aug 9;548(7666):E13-E14.
Tomasetti C, Vogelstein B. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 2015 Jan 2;347(6217):78-81.
Tomasetti C, Marchionni L, Nowak MA, Parmigiani G, Vogelstein B. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc Natl Acad Sci U S A. 2015 Jan 6;112(1):118-23.
Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa JP. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci U S A. 1999 Jul 20;96(15):8681-6.
Cho KR, Vogelstein B. Suppressor gene alterations in the colorectal adenoma-carcinoma sequence. J Cell Biochem Suppl. 1992;16G:137-41.
Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM, Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988 Sep 1;319(9):525-32.
Tomasetti C, Vogelstein B. On the slope of the regression between stem cell divisions and cancer risk, and the lack of correlation between stem cell divisions and environmental factors-associated cancer risk. PLoS One. 2017 May 16;12(5):e0175535.
Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983 Jan 6;301(5895):89-92