Lynch Syndrome
HEREDITARY
5/7/20247 min read
Lynch Syndrome is named after Professor Henry Lynch, a famous medical oncologist who worked in Creighton University in Omaha, Nebraska. He noticed that colon cancer, stomach cancer and uterine cancer tended to run in families. He noted dominant inheritance of the cancers and suggested that there was a gene causing this. He was right, but it took 30 years to prove it.
The Cause of Lynch Syndrome: hereditary DNA mismatch repair deficiency
When cells divide, the DNA has to replicate. One strand acts as a template while an enzyme (DNA polymerase) copies it and makes the new strand that goes into the daughter cell. Quite often the two strands slip on each other creating a mismatch. This means that the new strand doesn’t match the old one. A mutation has happened. Luckily there is a system of enzymes that pick up the mutation, cut it out, and allow the DNA polymerase to repair the new DNA. If this repair system were to fail there would be thousands of mutations in every cell. Some of these mutations would affect tumor suppressor genes and they would promote cancer.
This is the cause of Lynch syndrome; a germline mutation is one of the genes that repair the DNA mismatches. These genes are MSH2, MLH1, MSH6, and PMS2.
Defective DNA Mismatch Repair (dMMR)
dMMR is a hallmark of Lynch syndrome tumors and detecting it is a way to diagnose the syndrome. PCR analysis of tumor cells shows that the length of certain microsatellite markers in the DNA is different to that of the same markers in normal tissue. The microsatellites are “unstable”. If ≥20% of a set of 5 markers are unstable the instability is high (MSI-high). This is what is seen in Lynch cancers. 18% of colon and rectal cancers are MSI-high but only 3% are Lynch cancers. The commonest cause of MSI-high cancer is hypermethylation, caused by BRAF mutations in the colon which leads to a sessile serrated lesion. Then the hypermethylation shuts off expression of other genes including MLH1 and perhaps APC. The SSL becomes dysplastic and turns to cancer (MLH1) or becomes a TSA (APC) and turns to cancer.
A DNA Microsatellite is a length of DNA with repeated bases. There can be one base repeated (AAAAAAAAA), or two (TGTGTGTGTG) or three (GTAGTAGTAGTA) or even four (ATCAATCAATCAATCA). These are “slippery” bits of the gene which is why they need repair.
Diagnosing Lynch Syndrome
Lynch Syndrome, or hereditary DNA mismatch repair deficiency, is defined as the presence of a pathogenic (harmful) variant (or mutation) in one of the mismatch repair genes. Therefore it can only be diagnosed through genetic testing. Suspicion of Lynch Syndrome is an indication to test for the mutation, can be aroused by three scenarios.
1. Young age of onset of cancer. The average age of cancer diagnosis in Lynch Syndrome is 46. Much younger patients can be affected. Anyone with a colon or rectal cancer under the age of 50 needs genetic testing.
2. Strong family history. Because Lynch syndrome is dominantly inherited we expect cancer to run in the family. The Amsterdam Criteria (revised) were designed to detect dominant inheritance of cancer. They apply to a family and are:
i. Someone with colon or other Lynch syndrome cancer under the age of 50
ii. At least 3 people are affected over at least 2 generations
iii. 2 people are first degree relatives of the third
iv. Nobody has FAP (Lynch patients can have up to 20 polyps)
Patients with cancer whose family meets these criteria should get genetic testing.
3. Tumor Testing. For at least 13 years colorectal and some other tumors (endometrial) have been tested to see if there is evidence of defective mismatch repair. This done with MSI testing or Immunohistochemistry IHC). IHC stains tissue with an antibody against the protein we are interested in…with Lynch Syndrome there are 4 antibodies; anti MSH2, anti MLH1, anti MSH6 and anti PMS2. If a tumor doesn’t pick up the stain it means there is no corresponding protein in the tissue and that Lynch is likely. It is very likely if the proteins that are missing are MSH2, MSH6 or PMS2. If the missing protein is MLH1, it is more likely that a sporadic BRAF mutation and hypermethylation has caused the cancer. The tumor can be tested for the BRAF mutation and hypermethylation. If these are normal, then Lynch is likely. Abnormal IHC is followed by genetic testing.
Only about 10% of patients with colon cancer under age 50 will have Lynch. Only about half of those with an Amsterdam +ve family will have Lynch. IHC can be false positive. Difficult cases should be discussed with experts in the field including genetic counselors.
The Spectrum of Lynch Tumors
As with any dominantly inherited condition, the mutation causing the disease is present in every cell in the patient’s body. Certain tissues are vulnerable to the cancer-causing effects of the mutation and are at risk of getting cancer. In Lynch Syndrome there can be: gastric cancer, small bowel cancer, hepatobiliary tract cancer, urologic tract transitional cell cancer, ovarian cancer, skin cancer and brain tumors, in addition to colorectal and endometrial cancer.
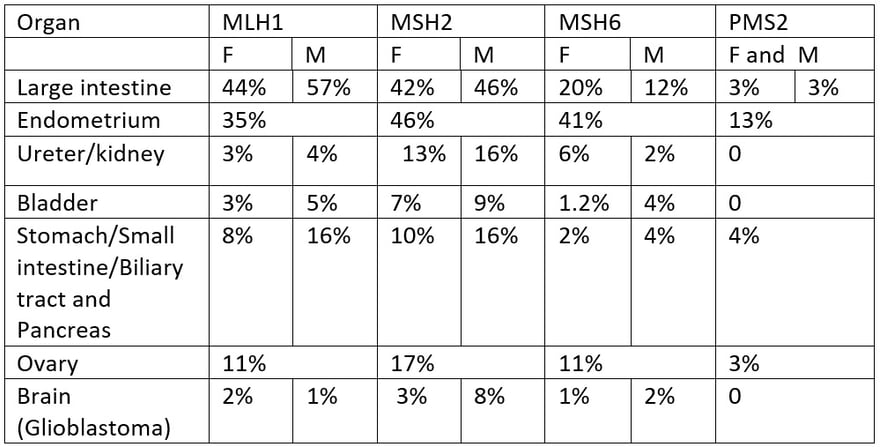
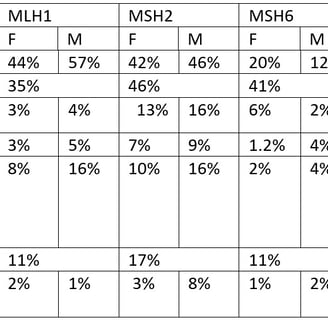
Risk of cancer at age 70 according to genotype


Kravochuck SE, Church JM. Colonoscopy in patients with Lynch syndrome: no room for error. ANZ J Surg 2017; 87: 204-205
Does the Gene Matter?
If you look at the table of cancer risk according to the gene carrying the mutation you will see that the most severe phenotypes are associated with MSH2 and MLH1 mutations. MSH6 mutations have a moderate cancer risk and PMS2 mutations have a very low cancer risk. The risk of cancer also varies by family, independent of gene. For example, some MSH2 families are severely affected with a high prevalence, while others are more mildly affected. We tailor treatment to the clinical pattern, always erring on the side of being too careful.
Constitutional Mismatch Repair Deficiency (CMMRD)
This is a condition where a patient has inherited two mismatch repair gene mutations from birth. This mean that a both parents had Lynch Syndrome. None of their cells can repair DNA mismatches ever. As expected they develop tumors in several organs at a very young age and usually die before they turn 20. The tumor spectrum includes café au lait spots on the skin, brain tumors (glioma), cancers in the blood and lymphatic system, polyps and cancers in the intestinal tract, uterine and ovarian cancers.
Screening for these cancers starts early. Although most cannot be prevented at least early diagnosis increases the chance of effective treatment.
Lynch Syndrome and the Immune Response
One of the first things that pathologists noticed about cancers in patients with Lynch Syndrome was evidence of an active immune response in the tumor. There was an increased number of tumor infiltrating lymphocytes (TILs) along with a Crohn’s –like inflammatory response. At the same time clinicians were noticing that Lynch Syndrome cancers had a better prognosis than the same cancers in patients without Lynch Syndrome. For years we wondered why.
Here is the answer:
The unstable microsatellites created by unrepaired DNA mismatches do not make normal proteins. But they do make chains of amino acids that are called frameshift polypeptides (FSPs). It turns out that these FSPs are very immunogenic. They stimulate the body’s immune system because they are “foreign”. This is a feature of all MSI cancers, including Lynch Syndrome cancers. This discovery matched the development of drugs that activate the body’s T cell response to cancers, such a pembrolizumab (Keytruda). Immune therapy is very effective in treating Lynch Syndrome cancers that have metastasized.
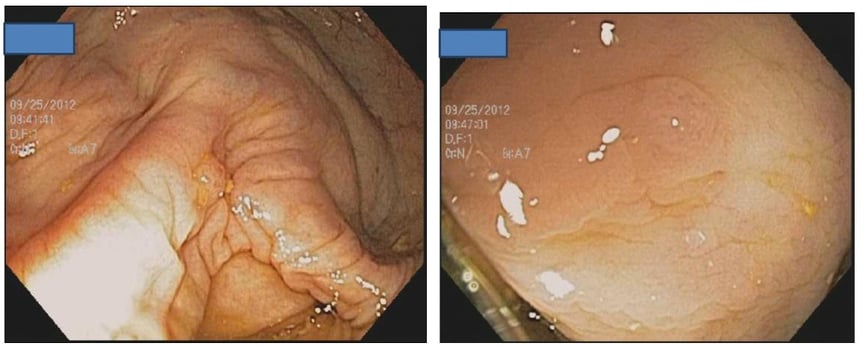
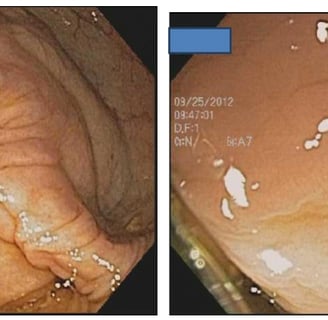
Colonoscopy in Patients with Lynch Syndrome
The polyp-cancer sequence in patients with Lynch Syndrome can be fast. Cancers can develop in the year between colonoscopies.
Therefore colonoscopy must be uncompromising:
Excellent preparation
Meticulous examination
Remove everything
Repeat yearly to two yearly
Two adenomas in a patient with Lynch Syndrome
Treatment can be discussed in four different contexts:
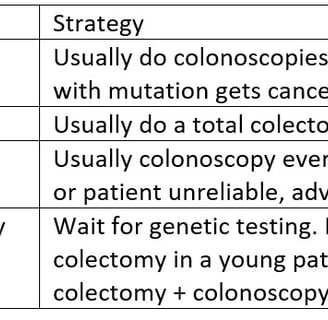
A gene mutation positive patient with no cancer
A gene mutation positive patient with a cancer before surgery
A gene mutation positive patient who has had a cancer removed before the mutation was known
A patient who has a positive family history suggesting Lynch who has a cancer
Dominguez-Valentin M, Sampson JR, Seppälä TT, Ten Broeke SW, Plazzer JP, Nakken S,et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the Prospective Lynch Syndrome Database. Genet Med. 2020 Jan;22(1):15-25.
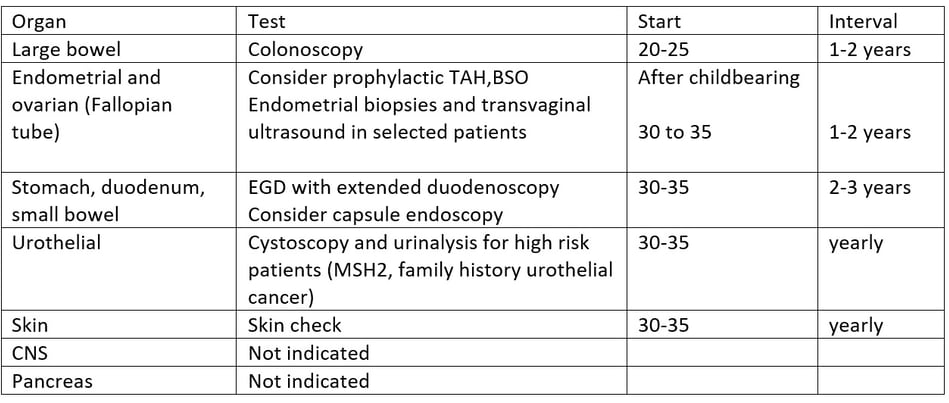
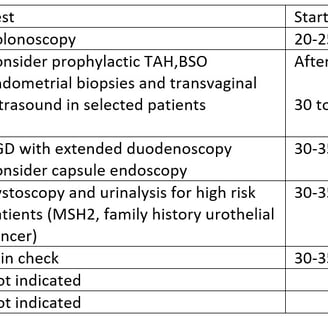
Surveillance recommendations adapted from NCCN
Treatment of Patients with Lynch Syndrome
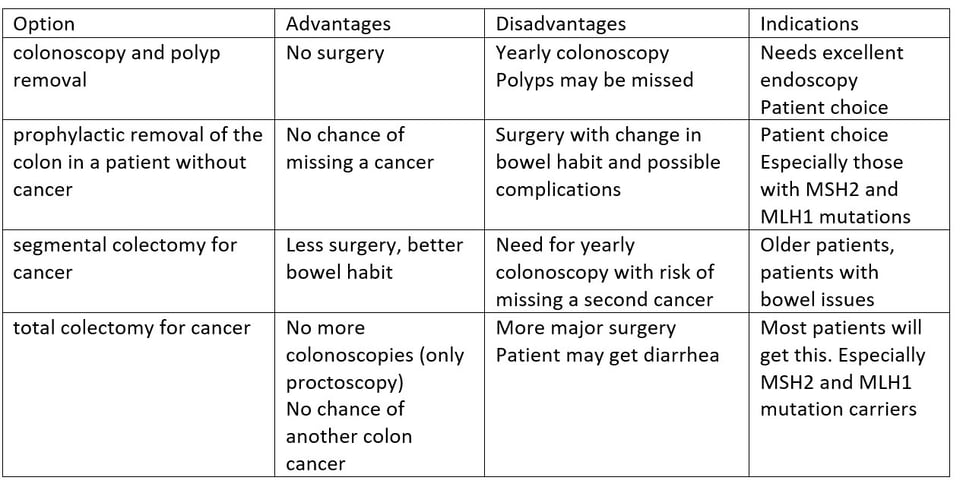
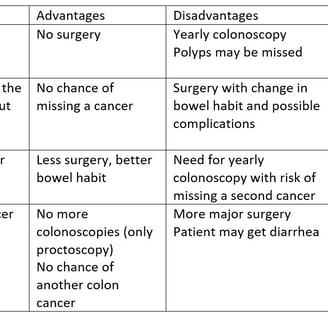
The primary risk in patients with Lynch Syndrome is Colorectal Cancer. This can be primary cancer, or metachronous cancer. Patients with one cancer are at high risk of developing another one and this risk determines surgical strategy. The options for treatment are: colonoscopy and polyp removal, prophylactic removal of the colon in a patient without cancer, segmental colectomy that would be done for patients without Lynch, total colectomy to remove the rest of the colon and prevent further cancers.
The advantages and disadvantages of the different options are in the table.