Hereditary Colorectal Cancer
HEREDITARY
5/30/202410 min read
Definition
Hereditary Colorectal Cancer includes all syndromes of cancer risk associated with inherited of a specific gene or genes and primarily affecting the large intestine. In addition we include patients diagnosed with colorectal cancer under the age of 50 and families exhibiting dominant inheritance of colorectal cancer without a positive genotype.


Diagnosis
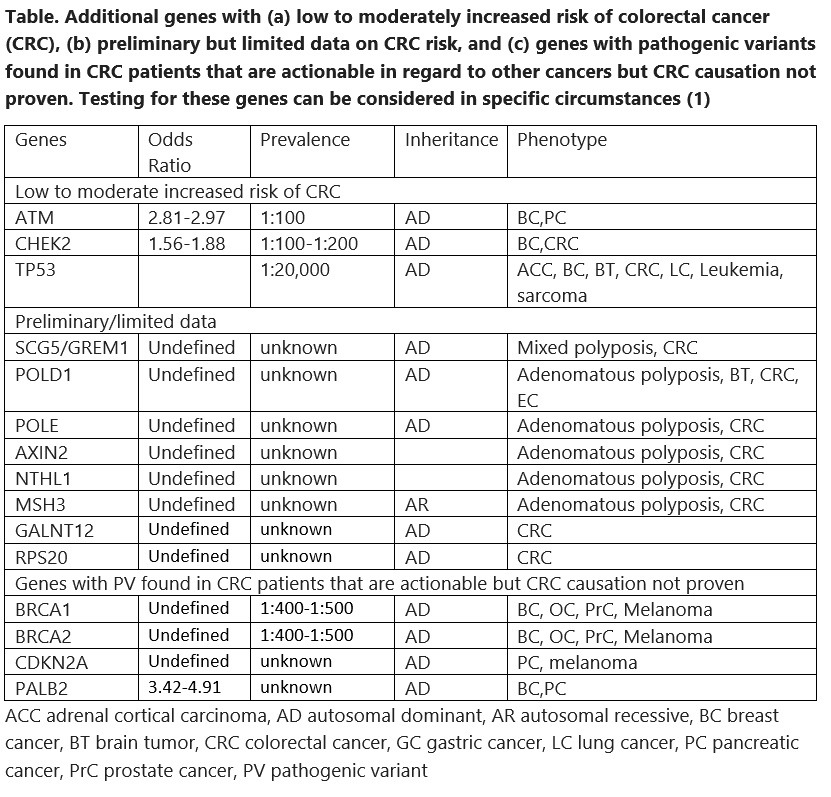
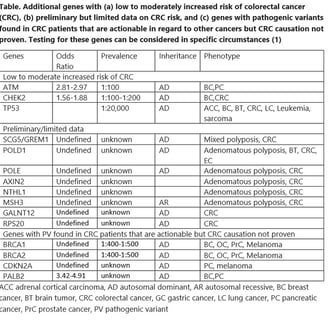
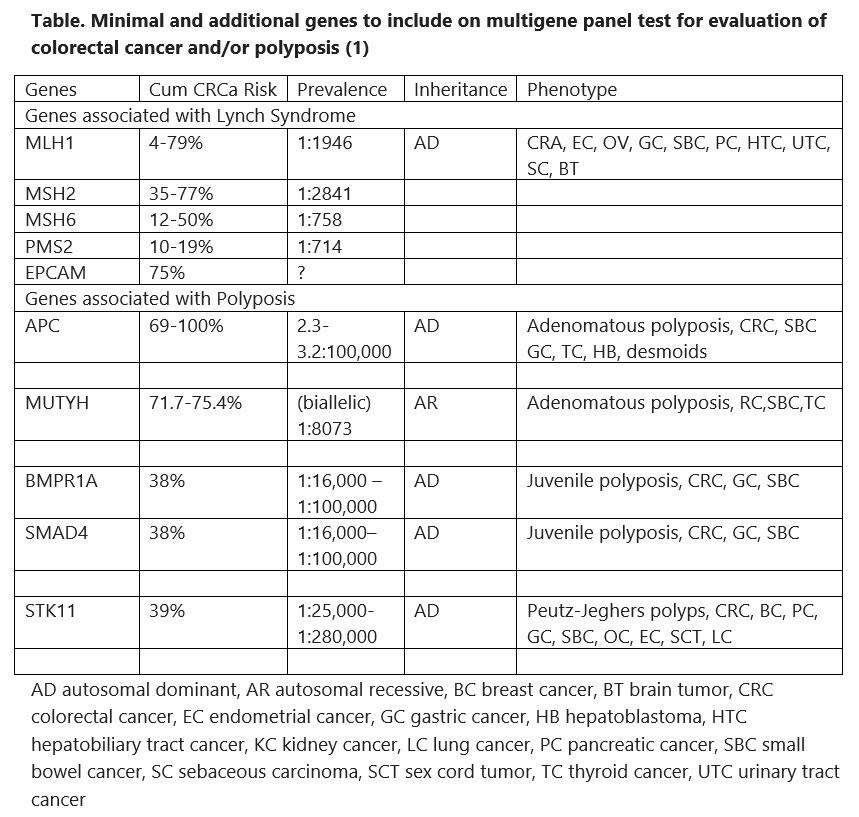
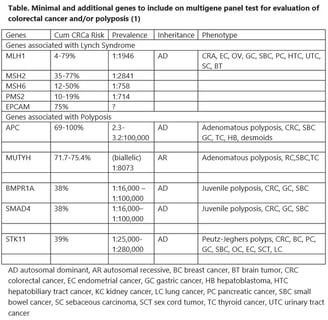
The foundation of care for a family with HCRC is diagnosis. For classical dominant syndromes with high penetrance this should be relatively easy. However prevalence varies, even with the same genotype. Furthermore, not every syndrome is dominantly inherited, and some families are small and do not show a convincing pattern of colorectal cancer. In general there is so much phenotypic overlap between HCRC syndromes that clinical diagnosis is suboptimal. Classical and profuse adenomatous polyposis is the clearest phenotype as there is no other syndrome associated with >100 colorectal adenomas. Table 1 summarizes the indications for panel testing recently recommended by the Collaborative Group of the Americas for Inherited Gastrointestinal Cancer (1). They are quite broad, based on the increased sensitivity of panel testing for finding a syndrome and the markedly decreased cost. Approximately 6% of average risk screening colonoscopies reveal 10 or more adenomas, and a cumulative count of 10 is much more common. About 10% of colorectal cancers are diagnosed in patients <50 years of age and the percentage is gradually increasing. About 9% of the population has a family history of a first degree relative with colorectal cancer. Most of the panels available for colorectal cancer risk include 25 to 40 genes that cover all the known syndromes and all the genes associated with non-syndromic moderate or even low colorectal cancer risk. Insurance coverage for some of the indications may vary and pretest counseling should include a discussion of this aspect.
Table 1. Collaborative Group of the Americas: recommended indications for multigene panel testing in the diagnosis of HCRC syndromes (1)
≥ 10 cumulative colorectal adenomas
Colorectal cancer diagnosed age < 50 years
Colorectal cancer and at least one first degree relative with colorectal or endometrial cancer
Mismatch repair-deficient colorectal cancer, not attributed to MLH1 promoter methylation
PREMM5 score ≥ 2.5% or MMRpro or MMRpredict score ≥ 5%
Multiple Lynch syndrome primary tumors
≥ 3 cumulative gastrointestinal hamartomatous polyps
Patients meeting any other genetic testing criteria
Lynch syndrome
The commonest of the HCRC syndromes is Lynch Syndrome, or hereditary mismatch repair deficiency. The Amsterdam Criteria, conceived to facilitate research, has been used as an indication for genetic testing, but only 50% of Lynch families are Amsterdam compliant. An Amsterdam compliant family is referred to as having Hereditary Non Polyposis Colorectal cancer (HNPCC), but HNPCC is NOT a synonym for Lynch syndrome. Lynch syndrome is defined by the presence of a pathogenic variant in one of 4 DNA mismatch repair genes in the germline of a patient.
PREM5 is a Lynch Syndrome prediction model based on age, gender, phenotype and family history. It is more accurate at predicting Lynch syndrome than the Amsterdam Criteria, but falls short of genetic testing.
Lynch syndrome, or hereditary DNA mismatch repair, is a specific condition due to a germline pathogenic variant in one of 4 DNA mismatch repair genes. DNA mismatch repair (MMR) corrects mismatches that occur during DNA replication at repetitive DNA sequences (microsatellites). Deficient DNA mismatch repair results in failure of repair of these mismatches and leads to microsatellite instability (MSI). MSI in a colorectal cancer is an indication of failed or deficient MMR (dMMR). Immunohistochemistry IHC) of a tumor caused by loss of function of a mismatch repair gene will show lack of staining for the protein produced by the gene, reflecting loss of expression of the gene itself. These tests, PCR for MSI and IHC, have become part of the standard analysis of colorectal cancers, and some of the larger colorectal adenomas. This is a screen for Lynch syndrome.
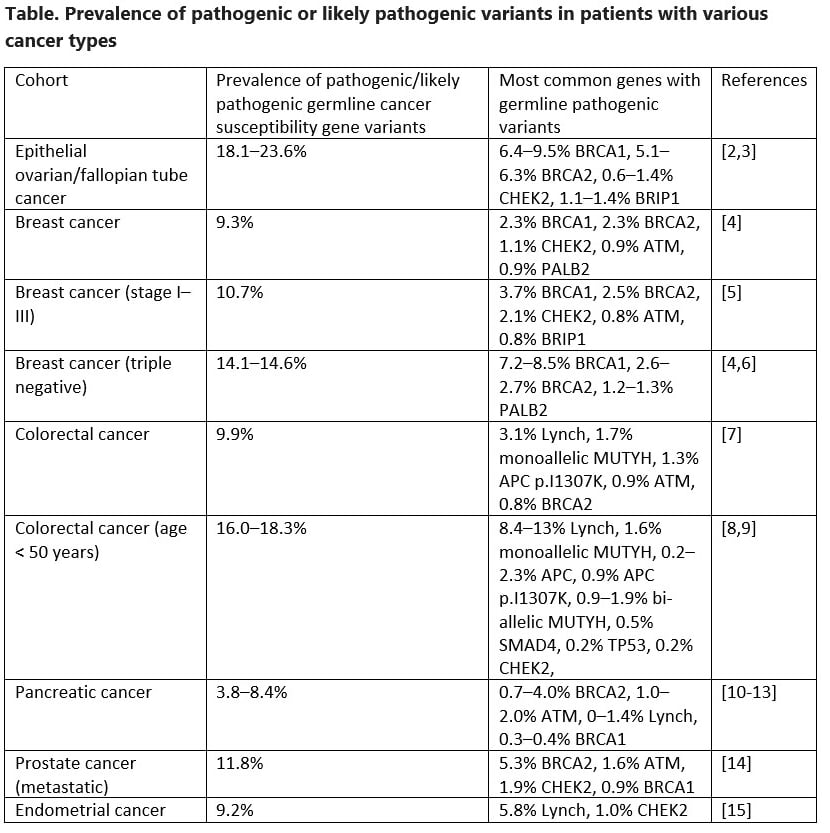
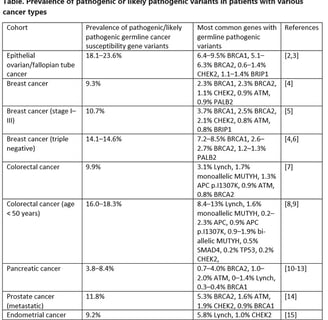
Some 15 to 18% of colorectal cancers exhibit MSI. Only 3% of colorectal cancer are due to Lynch syndrome. The most common cause of MSI in colorectal cancers is loss of the MMR gene MLH1, caused by DNA methylation. DNA methylation is a normal way for regulating gene expression. Increasing levels of methylation reduce gene expression. Lower levels of methylation increase gene expression. DNA methylation in the right colon increases with age, and a particularly high level of methylation is associated with pathogenic variants in the proto-oncogene BRAF. The pattern of hypermethylation associated with BRAF variants is known as CpG Island Methylator Phenotype (CIMP). CIMP produces serrated adenomas and leads to CIMP high, MSI cancers. CIMP is also involved in Sessile Serrated Polyposis. Most MSI colorectal cancers are CIMP related. When a colorectal cancer is shown to be MSI and MLH1 deficient, reflex testing for a BRAF variant and for hypermethylation are done. If there are no BRAF variants, and methylation levels are normal, the patient undergoes testing for MLH1 germline pathogenic variant (PV). If there is a germline PV the diagnosis is Lynch Syndrome. The family is counseled and tested. If there is no germline PV the tumor is “Lynch Like”. Sequencing of the tumor itself usually reveals biallelic somatic PVs in one of the MMR genes. In this case there is no inherited syndrome and the family is not tested for Lynch.
If IHC of a colorectal cancer reveals loss of expression of any of the other three MMR genes (MSH2, MSH6, PMS2) the diagnosis is Lynch syndrome and the family is counseled and tested.
The Family History:
Prior to genetic testing the family history was the only tool for accessing the majority of families with HCRC. A thorough family history (including age and cause of death for 3 generations) is required for a sensitive evaluation. Currently this has been incorporated into some electronic medical records programs but it can be done manually. Pedigrees can be drawn that summarize disease in the family and are helpful in guiding testing and surveillance. In the age of panel testing, pedigrees are still important to present a summary of the family.
The Registry:
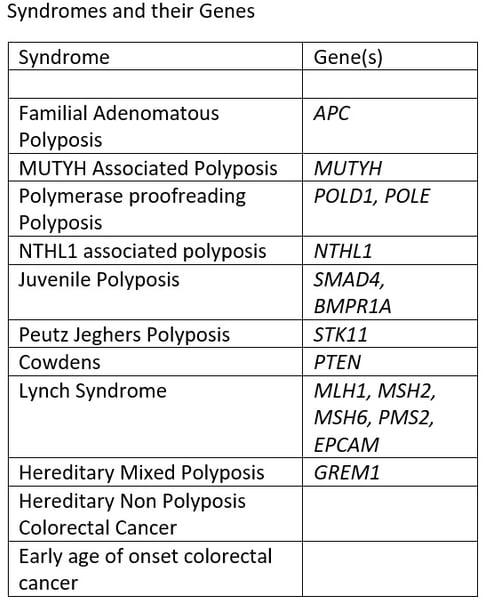
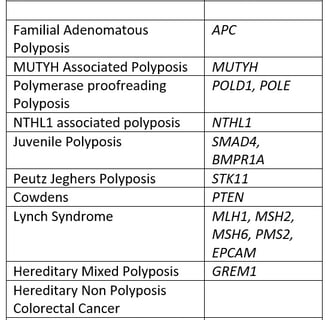
The three pillars of a center for HCRC are patient care, education and research. All are important and all depend on an effective database. Clinical use of a database includes the rationalization and organization of appointments and surveillance. Educational use allows the distribution of educational material, largely digital now, and the research role of the registry is to allow access to clinical data for use in various studies. There are currently at least 12 syndromes of HCRC, with different genotype and phenotypes. It makes sense to use different databases, each genotype specific: that is specifically designed for one or two syndromes that share a genotype. Examples of databases and fields are attached.
The Syndromes
Diagnosing Syndromes
A syndrome is a collection of disorders that are all typical of one cause, so that you can diagnose the cause by seeing one typical disorder. For example in Peutz Jeghers Polyposis, affected children get freckles on their lips. While this is obvious it is also rare. Most of the symptoms shown in the table cause polyps, and there is overlap between the symptoms. The best way to diagnosis is there for by getting genetic testing. Although we do not have the capacity to test everyones DNA for every gene we do have cheap genetic testing available that will rule out or diagnose most of these syndromes. The indications for testing and the process are shown in the next page.
The Process of Genetic Testing
Once the referral is made an appointment for pre test counseling with a genetic counselor is set. This is educate you about genetic testing, what it can do and what it can’t do. There are implications to genetic testing that may affect you socially or financially.
The test is done by collecting blood or saliva. There is a colorectal cancer panel that tests all the known genes associated with colon or rectal cancer. The results take 2 to 3 weeks. Then a post test visit to the counselor is set up to discuss the results.
Introduction
Hereditary Colorectal Cancer (HCRC) accounts for approximately 5% of all colorectal cancers. Patients and families affected by these syndromes are at high risk of colorectal and other cancers, occurring at an early age. This high level of risk can be managed by timely diagnosis and appropriate surveillance. Often the proband in the family is not screened and presents with one aspect of the disease phenotype, possibly cancer. Treatment of hereditary colorectal neoplasia is based on a therapeutic as well as a risk-reducing philosophy that takes into account the possibility of multiple lesions in several organ systems, and an unusually young age of onset.
Coordination of care of these patients and their families is optimally done using a system of documentation that records the genotype and phenotype of every person at risk. Affected individuals are then advised of their increased risk for neoplasia and a surveillance program is designed to detect, monitor and when necessary, to remove precancerous lesions, or early cancers that may have developed. Care sometimes involves multiple specialties, and education by counselors and coordinators. Combining appointments into one or two days minimizes inconvenience to the patients and in some syndromes where children are affected it is important to cater to the needs of the pediatric population.
All of these requirements can be met in a specialized center for Hereditary Colorectal Cancer. These centers can be found scattered in large medical centers around the country. There are several requirements for establishing such a center.
1. An interested and expert clinician. Most centers involve a colorectal surgeon and a dedicated gastroenterologist. In addition there are pathologists, radiologists, epidemiologists, statisticians, IT specialists and representatives of other medical specialties including General (upper GI, hepatobiliary) surgery, Obstetrics and Gynecology, Urology, Dermatology, Psychology, and Oncology.
2. A clinical coordinator is the heart of a registry, linking patients/families with clinicians. The coordinator facilitates appointments and hospital care, maintains and searches for medical records, arranges and checks on surveillance.
3. A registry or list of patients and families sorted by syndrome is important. Family trees are used to triage kindreds for counseling and testing, and are updated at the yearly visits.
4. A Genetic counselor is key to the diagnosis of a patient and the triage of the family. They perform pre and post-test genetic counseling, arrange genetic testing, and contribute to the resolution of confusing results.
References:
Heald B, Hampel H, Church J, Dudley B, Hall MJ, Mork ME, Singh A, Stoffel E, Stoll J, You YN, Yurgelun MB, Kupfer SS; Collaborative Group of the Americas on Inherited Gastrointestinal Cancer. Collaborative Group of the Americas on Inherited Gastrointestinal Cancer Position statement on multigene panel testing for patients with colorectal cancer and/or polyposis. Fam Cancer. 2020 Jul;19(3):223-239.
Risch HA, McLaughlin JR, Cole DE, Rosen B, Bradley L, Kwan E, Jack E, Vesprini DJ, Kuperstein G, Abrahamson JL, Fan I, Wong B, Narod SA (2001) Prevalence and penetrance of germline BRCA1 and BRCA2 mutations in a population series of 649 women with ovarian cancer. Am J Hum Genet 68(3):700–710. 10.1086/31878749
Sopik V, Phelan C, Cybulski C, Narod SA (2015) BRCA1 and BRCA2 mutations and the risk for colorectal cancer. Clin Genet 87(5):411–418. 10.1111/cge.12497
Walsh T, Casadei S, Lee MK, Pennil CC, Nord AS, Thornton AM, Roeb W, Agnew KJ, Stray SM, Wickramanayake A, Norquist B, Pennington KP, Garcia RL, King MC, Swisher EM (2011) Mutations in 12 genes for inherited ovarian, fallopian tube, and peritoneal carcinoma identified by massively parallel sequencing. Proc Natl Acad Sci USA 108(44):18032–18037. 10.1073/pnas.1115052108102
Tung N, Lin NU, Kidd J, Allen BA, Singh N, Wenstrup RJ, Hartman AR, Winer EP, Garber JE (2016) Frequency of germline mutations in 25 cancer susceptibility genes in a sequential series of patients with breast cancer. J Clin Oncol 34(13):1460–1468. 10.1200/JCO.2015.65.0747
Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, Garofalo A, Gulati R, Carreira S, Eeles R, Elemento O, Rubin MA, Robinson D, Lonigro R, Hussain M, Chinnaiyan A, Vinson J, Filipenko J, Garraway L, Taplin ME, AlDubayan S, Han GC, Beightol M, Morrissey C, Nghiem B, Cheng HH, Montgomery B, Walsh T, Casadei S, Berger M, Zhang L, Zehir A, Vijai J, Scher HI, Sawyers C, Schultz N, Kantoff PW, Solit D, Robson M, Van Allen EM, Offit K, de Bono J, Nelson PS (2016) Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med 375(5):443–453. 10.1056/NEJMoa160314412
Ring KL, Bruegl AS, Allen BA, Elkin EP, Singh N, Hartman AR, Daniels MS, Broaddus RR (2016) Germline multi-gene hereditary cancer panel testing in an unselected endometrial cancer cohort. Mod Pathol 29(11):1381–1389. 10.1038/modpathol.2016.135
Stoffel EM, Koeppe E, Everett J, Ulintz P, Kiel M, Osborne J, Williams L, Hanson K, Gruber SB, Rozek LS (2017) Germline genetic features of young individuals with colorectal cancer. Gastroenterology. 10.1053/j.gastro.2017.11.00424
Chow E, Lipton L, Carvajal-Carmona LG, Arthur G, Bhathal P, Kaur G, Jaeger E, Woodford-Richens K, Howarth K, Tomlinson I, Macrae F (2007) A family with juvenile polyposis linked to the BMPR1A locus: cryptic mutation or closely linked gene? J Gastroenterol Hepatol 22(12):2292–2297. 10.1111/j.1440-1746.2007.04989.x
Buys SS, Sandbach JF, Gammon A, Patel G, Kidd J, Brown KL, Sharma L, Saam J, Lancaster J, Daly MB (2017) A study of over 35,000 women with breast cancer tested with a 25-gene panel of hereditary cancer genes. Cancer 123(10):1721–1730. 10.1002/cncr.3049854
Couch FJ, Hart SN, Sharma P, Toland AE, Wang X, Miron P, Olson JE, Godwin AK, Pankratz VS, Olswold C, Slettedahl S, Hallberg E, Guidugli L, Davila JI, Beckmann MW, Janni W, Rack B, Ekici AB, Slamon DJ, Konstantopoulou I, Fostira F, Vratimos A, Fountzilas G, Pelttari LM, Tapper WJ, Durcan L, Cross SS, Pilarski R, Shapiro CL, Klemp J, Yao S, Garber J, Cox A, Brauch H, Ambrosone C, Nevanlinna H, Yannoukakos D, Slager SL, Vachon CM, Eccles DM, Fasching PA (2015) Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J Clin Oncol 33(4):304–311. 10.1200/JCO.2014.57.1414
Cancer Genome Atlas Research Network. Electronic address aadhe, Cancer Genome Atlas Research N (2017) Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 32(2):185–203. 10.1016/j.ccell.2017.07.007
Grant RC, Selander I, Connor AA, Selvarajah S, Borgida A, Briollais L, Petersen GM, Lerner-Ellis J, Holter S, Gallinger S (2015) Prevalence of germline mutations in cancer predisposition genes in patients with pancreatic cancer. Gastroenterology 148(3):556–564. 10.1053/j.gastro.2014.11.042
Norquist BM, Harrell MI, Brady MF, Walsh T, Lee MK, Gulsuner S, Bernards SS, Casadei S, Yi Q, Burger RA, Chan JK, Davidson SA, Mannel RS, DiSilvestro PA, Lankes HA, Ramirez NC, King MC, Swisher EM, Birrer MJ (2016) Inherited mutations in women with ovarian carcinoma. JAMA Oncol 2(4):482–490. 10.1001/jamaoncol.2015.5495
Hampel H, Panescu J, Lockman J, Sotamaa K, Fix D, Comeras I, LaJeunesse J, Nakagawa H, Westman JA, Prior TW, Clendenning M, de la Chapelle A, Frankel W, Penzone P, Cohn DE, Copeland L, Eaton L, Fowler J, Lombardi J, Dunn P, Bell J, Reid G, Lewandowski G, Vaccarello L (2007) Comment on: Screening for Lynch syndrome (hereditary nonpolyposis colorectal cancer) among endometrial cancer patients. Cancer Res 67(19):9603 10.1158/0008-5472